Steven-Johnson Syndrome: Types, Causes, How it Starts, Pictures
Stevens-Johnson syndrome (SJS) is a rare, but very serious skin peeling condition that is caused by an allergic reaction to medications or an illness. It’s usually a reaction to medication that starts with flu-like symptoms, followed by a painful rash that spreads and blisters. Then the top layer of affected skin dies, sheds, and begins to heal after several days. This condition affects 1 to 2 per million people each year.
Classification of Stevens-Johnson syndrome?
Stevens-Johnson syndrome is a medical emergency that usually requires hospitalization. Treatment focuses on removing the cause, caring for wounds, controlling pain, and minimizing complications as skin regrows. It can take weeks to months to recover.
Although several classification schemes have been reported, the simplest classification breaks the disease down as follows:
• Stevens-Johnson syndrome: A minor form of toxic epidermal necrolysis, with less than 10% body surface area (BSA) detachment
• Overlapping Stevens-Johnson syndrome/toxic epidermal necrolysis: Detachment of 10-30% of the BSA
• Toxic epidermal necrolysis: Detachment of more than 30% of the BSA

What are the signs and symptoms of Stevens-Johnson syndrome?
Typical prodromal symptoms of Stevens-Johnson syndrome are as follows:
• Cough productive of a thick, purulent sputum
• Headache
• Malaise
• Arthralgia
Patients may complain of a burning rash that begins symmetrically on the face and the upper part of the torso. The cutaneous lesions are characterized as follows:
• The rash can begin as macules that develop into papules, vesicles, bullae, urticarial plaques, or confluent erythema
• The typical lesion has the appearance of a target; this is considered pathognomonic
• In contrast to the typical lesions of erythema multiforme, these lesions have only 2 zones of color
• The lesion’s core may be vesicular, purpuric, or necrotic; that zone is surrounded by macular erythema
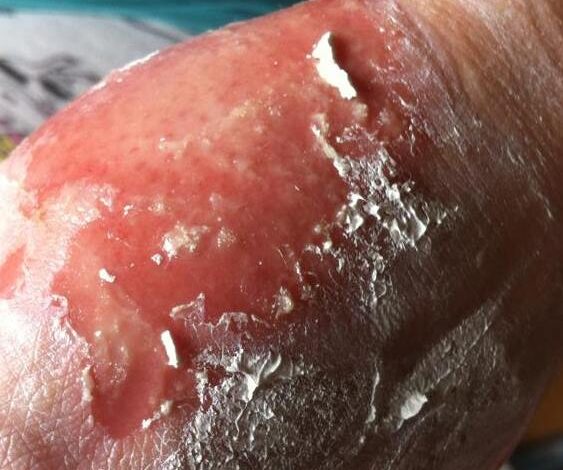
• Lesions may become bullous and later rupture, leaving denuded skin; the skin becomes susceptible to secondary infection
• Urticarial lesions typically are not pruritic
• Infection may be responsible for the scarring associated with morbidity
• Although lesions may occur anywhere, the palms, soles, dorsum of the hands, and extensor surfaces are most commonly affected
• The rash may be confined to any one area of the body, most often the trunk
Signs of mucosal involvement can include the following:
• Erythema
• Edema
• Sloughing
• Blistering
• Ulceration
• Necrosis
The following ocular signs may be noted on slit-lamp examination:
• Eyelids: Trichiasis, distichiasis, meibomian gland dysfunction, blepharitis
• Conjunctiva: Papillae, follicles, keratinization, subepithelial fibrosis, conjunctival shrinkage, foreshortening of fornices, symblepharon, ankyloblepharon
• Cornea: Superficial punctate keratitis, epithelial defect, stromal ulcer, neovascularization, keratinization, limbitis, conjunctivalization, stromal opacity, perforation (see the image below)
Severe damage to the skin and mucous membranes makes this condition a life-threatening disease. Because the skin normally acts as a protective barrier, extensive skin damage can lead to a dangerous loss of fluids and allow infections to develop. Serious complications can include pneumonia, overwhelming bacterial infections (sepsis), shock, multiple organ failure, and death. About 10 percent of people with Stevens-Johnson syndrome die from the disease, while the condition is fatal in up to 50 percent of those with toxic epidermal necrolysis.
What drug causes Steven-johnson syndrome?
There are several drugs and medications that can trigger Stevens-Johnson syndrome. Genetic changes have been found to increase the risk of Stevens-Johnson syndrome in response to triggering factors such as medications. Most of these changes occur in genes that are involved in the normal function of the immune system. The list of drugs and medications that can cause Stevens-Johnson syndrome include:
• Abacavir
• Allopurinol
• Antifungals
• Antivirals
• Atenolol
• Azathioprine
• Beta-lactam antibiotics
• Captopril
• Carbamazepine
• Cephalosporins
• Clomipramine
• Dapsone
• Diltiazem
• Gold salts
• Imidazole antifungals, eg ketoconazole, itraconazole, fluconazole
• Isoniazid
• Mexiletine
• Minocycline
• Naproxen
• Nevirapine (non-nucleoside reverse-transcriptase inhibitor)
• Nonsteroidal anti-inflammatory drugs (NSAIDs)(oxicam type mainly)
• Paracetamol
• Penicillins
• Phenobarbitone
• Phenytoin
• Sulfasalazine
• Sulfonamides
• Trimethoprim
• Valproic acid
• Vemurafenib
Are there other factors that increase the risk of someone developing Stevens-Johnson syndrome (SJS)?
You are at greater risk of SJS if you have the following conditions:
• Bone marrow transplant.
• Systemic lupus erythematosus.
• Human immunodeficiency virus (HIV).
• Other chronic diseases of joints and connective tissue.
• Cancer.
• Weakened immune system.
• Family history of SJS.
• Variation of a specific gene called human leukocyte antigen-B.
How quickly does Steven Johnson syndrome spread?
Symptoms of drug-induced Steven Johnson syndrome appear about one to three weeks after you start taking medication. The flu-like illness (fever, cough and headache, skin pain) is followed first by a rash and then peeling. In the case of TEN, some people even lose hair and nails.
Is Steven-johnson syndrome contagious?
No, Steven-johnson syndrome is not contagious, it is an unpredictable adverse reaction to certain medications. It can also sometimes be caused by an infection. The syndrome often begins with flu-like symptoms, followed by a red or purple rash that spreads and forms blisters.
How is Steven-johnson syndrome treated?
Treatment of Steven-johnson syndrome is most successful when Stevens-Johnson syndrome and toxic epidermal necrolysis are recognized early and treated in an inpatient dermatologic or intensive care unit setting; treatment in a burn unit may be needed for severe disease. Ophthalmology consultation and specialized eye care are mandatory for patients with ocular involvement. Potentially causative drugs should be stopped immediately. Patients are isolated to minimize exposure to infection and are given fluids, electrolytes, blood products, and nutritional supplements as needed. Skincare includes prompt treatment of secondary bacterial infections and daily wound care for severe burns. Prophylactic systemic antibiotics are controversial and often avoided.
Drug treatment of Stevens-Johnson syndrome and toxic epidermal necrolysis is controversial. Cyclosporine (3 to 5 mg/kg orally once/day) inhibits CD8 cells and has been shown to decrease the duration of active disease by 2 to 3 days in some instances and possibly decrease mortality. The use of systemic corticosteroids remains controversial. It had been thought by many experts to increase mortality because of increased rates of infection and the risk of masking sepsis. However, some reports show improved outcomes with early corticosteroid therapy.
Plasmapheresis can remove reactive drug metabolites or antibodies and can be considered. Early high-dose IVIG 2.7 g/kg over 3 days blocks antibodies and Fas ligand. However, despite some remarkable initial results using high-dose IVIG for toxic epidermal necrolysis, further clinical trials involving small cohorts have reported conflicting results, and a retrospective analysis has suggested no improvement or even higher than expected mortality.
The TNF-alpha inhibitors infliximab and etanercept can help reduce inflammation. Thalidomide has also been tested but increases mortality and is now contraindicated.